Lizzy is one of our recent PhD graduates from the CHOICE Institute (Class of 2020). We are very excited to have Lizzy share her experience and tips with us!
“I found that a methodical, small-steps approach helped me not feel overwhelmed by whole process.”
Lizzy Brouwer
Why did you choose to do a PhD? And how did you choose health economics?
I fell into health economics as a way to merge my two loves as an undergraduate student: social policy and mathematics. I was interested in researching and promoting policies that increased equitable access to affordable health care, which lead me to a Master of Public Health in health policy at the University of Michigan in Ann Arbor. We were required to take several economics courses as part of the curriculum, and I loved applying economic principles to real-world data in order to answer health policy questions. After a few years in research, I realized I wanted to dive deeper into the methods I was using and eventually take a bigger leadership role in projects. That led to a PhD program.
What was the topic of your PhD?
My dissertation was titled, “Exploring the uptake of value-based formulary strategies and their application to specialty drugs.” My first aim explored whether patient out-of-pocket costs were associated with value as defined by cost-effectiveness analysis to understand the integration of value information in private formularies. My second aim looked at the use of patient assistance programs (also commonly referred to as copay coupons) for specialty drugs and how their presence affected patient demand. The purpose of this aim was to understand if patients were responsive to changes in out-of-pocket costs for specialty drugs in the face of manufacturer-provided financial assistance, and therefore whether value-based insurance design could be an effective lever in that context.
What are you currently working on? Does this align with your training or your research interests?
I am currently working in two jobs. The first is as a research scientist at CHOICE building models in collaboration with the Institute for Clinical and Economic Review (ICER). This exciting opportunity allows me to be involved in national conversations about drug value with one of the leading health economics organizations in the United States, with the added bonus of continuing to work with the wonderful CHOICE faculty. My other role is a research associate with Curta Consulting, which was founded by CHOICE alumna, Lisa Bloudek. In this position, I work on research projects and model building for various biopharmaceutical companies. I really enjoy the fast pace and the wide variety of topics covered, as well as better understanding the role and nuances of health economics in the private sector.
Can you share one of your favorite or proudest moments during the PhD years?
I had my first child during my 3rd year, and it was harder than I anticipated to manage my family responsibilities while also moving my dissertation forward. When I returned from maternity leave at the beginning of fall quarter, I was determined to regain any lost momentum and I created an ambitious timeline for myself. Therefore, my proudest moment of the program was in the spring of that academic year when I had my long dissertation proposal approved, my general exam scheduled, and was awarded both the Lou Garrison Award for work in Health Economics and the PhARMA Foundation Predoctoral Fellowship in Health Outcomes. It felt like validation for my commitment to momentum during the previous months.
I also scheduled regular meetings with my chair and printed out an itemized agenda for each meeting to ensure our time was used efficiently… By writing everything down and methodically checking it off, I was able to feel reasonably sure I was not dropping any of the details I was juggling.
What do you think are the “secret sauces” of a successful PhD experience?
Every student is different and will have different paths to success, however I found that a methodical, small-steps approach helped me not feel overwhelmed by whole process. This meant outlining major milestones and then breaking down all the steps needed to get there. Once the larger tasks were broken down, they felt much more manageable. For example, I created deadlines for myself to produce and submit drafts of my dissertation products (short proposal, long proposal, analysis plans, manuscripts, dissertation chapters, etc), and I stayed (mostly) accountable to those deadlines. I also scheduled regular meetings with my chair and printed out an itemized agenda for each meeting to ensure our time was used efficiently. For example, if there was a roadblock in my research, I tried to outline the problem and suggest 2-3 paths forward for us to discuss. By writing everything down and methodically checking it off, I was able to feel reasonably sure I was not dropping any of the details I was juggling.
Advice for current/ future PhD students?
You can do it! Take breaks, have fun, and finish the darn thing 😊
We are starting a series of interviews with graduates from the CHOICE Institute, and are very excited to have Nathaniel Hendrix, who graduated from the PhD program in 2020, share his experience with us.
Nathaniel in the grad room, before the pandemic
“Grad school is like training a pet, where the pet is your own mind… You have to be intentional about figuring out what you want to teach yourself and how you’re going to do it.”
Nathaniel Hendrix
Why did you choose to do a PhD? How did you choose health economics?
I entered the PhD program directly after graduating with my PharmD. I went into my clinical training imagining that almost all of our decisions were grounded in a solid basis of evidence. But once I learned to read studies for myself, I began to see how complex the processes of gathering and evaluating evidence really were. This made me a bit hung up on how clinicians make decisions and how they can learn to make them better.
I had always been pretty indulgent with myself about taking electives during my PharmD training. I’d explored computer science and philosophy of science, but when I started taking classes in health economics, that gave me a vocabulary for talking and thinking about decision-making that I hadn’t been able to find before. At the same time, it’s a relatively young field and a very interdisciplinary one, so it felt to me like I could impact the direction of the field and that my curiosity about exploring different ideas would be rewarded.
2. What was the topic of your PhD?
My dissertation was on using health economics tools to solve translational issues in artificial intelligence (AI). It had two separate aims. First, I conducted a discrete choice experiment to see what primary care providers see as valuable in how AI could be used for breast cancer screening. Most women who take part in breast cancer screening make the decision to start with their primary care providers, so these providers have an outsized role to play in determining how AI will be used for breast cancer screening. We found that there were multiple ways that developers can appeal to primary care providers with their AI products: by improving sensitivity, by having a well-thought-out workflow that includes radiologists, and by having highly diverse training data.
The second aim had to do with using cost-effectiveness analysis to inform how AI algorithms are used for breast cancer screening. We used a real set of AI algorithms that had been submitted to a contest and used different methods of selecting a sensitivity/specificity threshold at which they could operate. Our model ended up selecting very similar thresholds as other heuristic methods, but it taught me a lot about the challenges of using cost-effectiveness analysis on AI.
“Write down every research idea that you have, and revisit that list frequently. Half of the ideas will be terrible in retrospect, but that’s okay.”
3. What are you currently working on? Does this align with your training or your research interests?
After my PhD, I started a postdoc at the Harvard T.H. Chan School of Public Health, where I’m mostly studying cost-effectiveness methodology. I have three main projects right now. First, I’m working on developing methods for integrating financial risk protection into decisions about prioritizing healthcare interventions in low- and middle-income countries. Healthcare plays a major role in keeping people out of poverty, but this isn’t acknowledged in conventional cost-effectiveness analysis.
Next, I’m working on methods for assessing the cost-effectiveness of health system strengthening interventions, such as building new facilities, training personnel, or developing a new informatics infrastructure. This is challenging because these long-term projects, which almost everyone acknowledges as important, have to compete for funding against urgent needs like making more medicines available or expanding vaccinations.
Finally, I’m completing a project about how to use Deep Learning-based breast cancer risk scores to personalize screening. Even though breast cancer screening reduces cancer mortality, there are many downsides because the false-positive rate is pretty high in screening. So we’re hoping we can use AI to determine who is most likely to benefit from screening and who might have a higher risk of being harmed.
I see this last project as most connected to my PhD research, because I’m using many of the same techniques that I used in the second aim of my dissertation. But I’m also learning many methods like constrained optimization that aren’t emphasized at UW but that are important when thinking about operating a healthcare system efficiently. Of course, I’m also wrapping up several projects I started during my time at UW, so there’s some continuity there too.
4. What are some of the things you wish you’d known when you started your PhD?
I wish I had had a better system for keeping track of the things I was reading. Ultimately, I missed the opportunity for making connections because it took me until I was well into my dissertation to discover the tools I needed for taking notes and helping myself to rediscover ideas I’d read in the past. For me, this ended up being the Zettelkasten system, which I use on the website Roam Research (read Sönke Ahrens’s book, “How to take smart notes,” for more info on this!).
5. Can you share one of your favorite or proudest moments during the PhD years?
Probably my proudest moment was when I found out that I had gotten a PhRMA Foundation fellowship for my dissertation. I was on vacation in San Francisco, waiting out an afternoon rainstorm in a café when I got the call, and it was really a pivotal moment for me. I had been feeling a bit of imposter syndrome about my dissertation, but to know that these other researchers had found my ideas exciting enough to fund was a huge boost of confidence.
“I’m convinced that you can’t be a good researcher without learning to be a good reader.”
6. What do you think are the “secret sauces” of a successful PhD experience?
Completing a PhD program is a great chance to explore. I didn’t quite understand this when I went into it and felt pressured (by myself, really!) to focus more on one area or another. Once I started just saying yes to every experience I had time for, I started to learn a lot more. This meant not just learning more about different technical matters, but also learning about what sort of work styles and communication styles I’m most comfortable with. That’s super important, because our work is usually so collaborative.
As you go through a PhD program and learn more, what’s expected from you also changes. In the beginning, you’re soaking up new methods and concepts, so you end up doing a lot of work on tasks like data cleaning or literature review that are really time consuming but necessary. With more experience, you should be able to start making suggestions to your collaborators about the directions for your work. And then finally, with your dissertation, you get a lot more independence about its direction.
Part of this development process is having good reading habits. That means keeping track of what journals are publishing so that you have a sense of what current conversations are happening. And then, again, finding ways to make connections between what you read. Ultimately, I’m convinced that you can’t be a good researcher without learning to be a good reader.
7. What do you think is the best metaphor for your grad school experience? How would you complete this sentence: “Grad school is like…”
Grad school is like training a pet, where the pet is your own mind. During grad school, you’ll have to teach yourself a lot of new skills, so it’s important to think about how to keep yourself motivated. It’s also vital to learn to recognize when you’re tired and need to do something entertaining. But overall, you have to be intentional about figuring out what you want to teach yourself and how you’re going to do it.
8. Do you have any other advice for current/future PhD students?
Write down every research idea that you have, and revisit that list frequently. Half of the ideas will be terrible in retrospect, but that’s okay. It’s easier to come up with good ideas if you come up with lots of ideas. And, again, to come up with ideas, it’s important to cultivate a very intentional habit of reading academic literature.
Also, I would encourage students to publish broadly. Look at the areas where they’re doing projects, compare it to the major areas in the CHOICE curriculum, and see if there are any weaknesses. You’ll have a lot more flexibility with your job search if you publish broadly. One way to do this is to collaborate with a lot of different people who are working in different areas.
From left to right: Brennan Beal, Joyce Jiang, Yilin Chen, Jacinda Tran, Sara Khor, Charles Phelps
The coronavirus disease 2019 (COVID-19) pandemic has shone a spotlight on the shortcomings of the current US health care system. There is an urgent need to address the problems with healthcare access, costs, and equity. Conversations related to health care reform have been and will continue to be front and center in the upcoming presidential election. Many health care reform proposals have been put forward, including the expansion of the Affordable Care Act, Medicare-for-All, and Medicare expansion that maintains a role for private insurers.
Earlier this year, Dr. Charles Phelps, a renowned health economist and the author of the Health Economics textbook, visited the CHOICE Institute and gave a lecture on his proposed health care plan: Medicare-Advantage-for-All. Under this proposal, all permanent residents will be able to choose among a wide variety of private insurance plans, similar to the Medicare Advantage program that is currently available to the older US population. All individuals will have health care coverage, with the minimum coverage being a high-deductible health plan with a health savings account. The health savings account can be filled up based on income levels. High value care such as preventative medicine can bypass the deductible.
We had the honor of sitting down with Dr. Phelps to talk more about his proposal.
Part 1 – On health care costs:
What are the most important drivers of the current cost growth in the healthcare system?
Dr. Phelps identified two main drivers of the healthcare cost growth: introduction of new technologies and the aging population. In the long run, he said, the introduction of new technologies, which includes diagnostic tools, pharmaceuticals, and genetic-based medicine, will increase costs. As treatment and drugs become more targeted, they will be sold in smaller quantities for focused populations. While these new advances will produce a lot of value, they will also drive cost growth.
As the size of the older population continues to expand, the age distribution pyramid (where the bottom is the youngest age group and the top is the oldest) will be shaped more like a cylinder. “When you look 20 years from now, this cylinder is going to have a great big hat on,” says Dr. Phelps. “The widest group on this cylinder… is going to be the oldest group at the top”.
This has important implication for healthcare costs, because the current way we are financing Medicare is through the payroll tax. The ratio of workers (who pay the payroll tax) to retired individuals (who use Medicare) is shrinking. “At the beginning of Medicare, this ratio was 4.5,” says Dr. Phelps. “In a couple of decades, this ratio is going to be 2 workers per retiree. The payroll tax mechanism of paying for Medicare has to change”.
How would the Medicare Advantage-for-All Plan address these cost drivers?
In a single payer system, usually one agency (e.g. CMS in Medicare) makes the determination about which new technology to introduce into the system. “They can make mistakes both by being too generous, letting too many things in the door, or being too stingy,” said Dr. Phelps. According to Dr. Phelps, the current US Medicare system is too generous, as it has not built in any cost constraints, while the British National Health Service has very tight cost-effective criteria for approval of new technologies.
The Medicare-Advantage-for-All plan would allow different plans to make cost-effectiveness evaluations. The exact details of the plan still need to be decided, but Dr. Phelps said that technologies that are very cost-effective, ICER between $50,000 to $75,000/quality-adjusted life year (QALY) gained, will be mandatory in the basic plan, and others will be left to the discretion of the insurance plan to cover it. “People will buy insurance plans to fit their needs, just like they buy different cars with different degrees of safety and pizzazz.”
What is the potential cost impact of Medicare Advantage for all? How is this cost impact compared to Medicare for All?
“Medicare-for-All is ghastly expensive, not because it offers universal health coverage, but because it eliminates all co-pays.” The RAND Health Insurance experiment and the Oregon Health Insurance experiment have demonstrated that medical use will increase when there is no co-pay or deductible. Dr. Phelps worried that the lack of co-pays will “unleash the monster in the evolution of new technologies,” referring to how moral hazard— the price sensitivity of demand for health care – may incentivize the use of low value care and technologies. He was concerned that healthcare costs will continue to rise unless there is a very tight constraint on new technologies.
In a way, Medicare-Advantage-for-All is similar to Medicare-for-All if everyone’s health saving account is filled up. When determining how much should be in the health saving accounts, Dr. Phelps said that “it will be an experiment through time to trade off the risk bearing and cost control,” adding that essential medicines and services, such as birth control or insulin for diabetes, can completely bypass the deductibles and co-pays.
Right now, Dr. Phelps said it is unclear how the different program parameters should be set in order to balance costs, new technology introduction, and equity considerations, but he is trying to devise an instrument that would allow users the flexibility to tweak parameters over time.
How about administrative costs? Dr. Phelps said that single payer plans, like Medicare-for-All, definitely reduce administrative costs. He argued that this administrative cost, although pricey, is giving us choice, and is controlling the costs by negotiating with providers about how much to pay for things instead of having fixed fee schedules. “Our society really values choice,” Dr. Phelps added. A one-size-fits-all plan takes away choice. Using an analogy in car shopping, Dr. Phelps said having just one healthcare plan is like saying: “You can buy any car you want, as long as it is a Honda Accord.”
Instead of simply comparing the administrative costs between a single-payer government-funded plan and Medicare-Advantage-for-All plans, Dr. Phelps emphasized the importance of taking into account the welfare loss and tax distortions that arise from the increase of taxation in a single-payer government plan. “Every dollar of tax we collect distorts the economy in some fashion…If you raise income tax, you change the labor supply.”
Part 2 – On choices and health technology assessment (HTA):
If there are many Medicare-Advantage-for-All health plans and each plan uses a different threshold, how do consumers make informed decisions about which plans to choose?
“Advisors will emerge,” said Dr. Phelps. Like the advisors for buying automobiles or financial advisors for the stock market, Dr. Phelps believed that a service for advising people on healthcare insurance and utilization will develop, and so would competition. These advisors could be independent or affiliated with big health plans. The emergence of these advisors, he added, will “depend on having access to electronic medical records that people can share.”
What role will HTA play? What role would an organization like the Institute for Clinical and Economic Review (ICER) play?
Dr. Phelps felt that the current way of conducting cost-effectiveness analyses is incomplete. Individuals who want to maximize their own utility do not necessarily think about everything that the society thinks about. While the QALY is a very important component of the overall value index, Phelps argued that the value index should include other things that are not necessarily built into a single individual’s utility, such as the fear of contagion and equitable distribution of health services. “Policies about Ebola, Zika, and now the coronavirus, are not made on the cost-effectiveness of vaccines. People would pay anything to get a coronavirus vaccine right now. The fear of contagion dominates public discourse and public policies. That is not captured in cost-effectiveness analysis.”
Ideally, Dr. Phelps says, there will be competition among HTA organizations to produce estimates with quality control by the government, similar to how the FDA has control over the new drugs that come on to the market. “I can see different insurance plans, [especially] some of the large ones, creating their own HTA shops. ICER would continue and offer [assessments] to smaller insurance plans.”
Part 3 – On equity:
Under Medicare-Advantage-for-All, multiple plans with varying premiums and deductibles mean that people who are better at navigating the system (e.g. more able to afford an advisor) or can afford the higher premiums may get more comprehensive care, potentially creating a situation where there is differential access and differential outcomes across the population based on education or wealth. How should we think about this?
“A plan that has absolutely equal access to health care…is imaginary”. Dr. Phelps argued that as long as people have different incomes, there would be differential care. Even under the British National Health System, those who can pay more can seek additional or better care with private insurance. In Ontario, Canada, where there is universal health coverage, some Canadians opt to purchase insurance to buy medical care in the U.S. One thing that the Medicare-Advantage-for-All plan can guarantee, Dr. Phelps added, is minimal level of access to quality care for everyone. The minimal plan could include an independent advisor.
How will international students be included in the plan?
International students are often in the U.S. for a few years. They are not permanent residents, but they are also not temporary visitors. It will be important that there are ways for these individuals to get access to the health plans. Some suggestions that Dr. Phelps had were to charge individuals an actuarially-based fee to join the plans, or to negotiate bilateral exchanges with different nations.
Part 4 — On political economy:
What are some of the potential political pushbacks related to this proposal?
The country is very heavily divided between those who want a single-payer plan (e.g. Medicare-for-All) and those who want to continue with private insurance. Those who are proponents of single-payer plans will not like Medicare-Advantage-for-All because it continues to use, as a central feature, and quite deliberately, private insurance plans. In this political climate, Dr. Phelps said, “My forecast would be…that there is zero probability that a Medicare-for-All plan would pass through the congress.”
“The high deductible health plan creates some anxiety among some people…for two reasons”, he continued. “One is that they say it discriminates against the poor.” Dr. Phelps said he can completely eliminate this concern by filling up the health savings accounts on an income-related basis. Another concern is that people in the high deductible plans will stop using the care that they need. A short run fix, Dr. Phelps explained, is that all highly-valued services and medicines, like diabetes medications, bypass any deductible and copayment.
“The long run fix is to vastly repair our vulnerable K-12 education system,” said Dr. Phelps. Higher education helps people navigate the highly complex health care system. There is also a very steep education gradient on lifestyle choices that are bad for your health, like tobacco smoking and binge drinking.
The COVID-19 pandemic has boosted the interest for mathematical models of infectious diseases. In this entry, I will briefly introduce some of these models and provide an R-code to simulate an outbreak of COVID-19.
These models synthesize multiple sources of information into equations that aim to model the evolution of a disease and make predictions. When used correctly, they can be incredibly powerful tools to explain a very chaotic and complex reality, to evaluate policy options to inform decision-making, to understand hidden mechanisms that drive an epidemic, and others.
Infectious diseases do not occur in isolation in each person. They are transmitted through contact with a pathogen. Thus, there is a need to understand the mechanisms for a susceptible person to establish effective contact (i.e. contact that results in a transmission; sexually transmitted disease is a good example) with someone who is infected with that pathogen. On the population level, disease prevalence is considered a risk factor for the incidence: the higher the proportion of people living with a disease, the higher the likelihood that an infected person gets in contact with a susceptible person. This relationship between incidence and prevalence can be characterized using dynamic models. Here, the probability of getting infected is determined by the probability of contact with an infectious person (or animal in case of diseases transmitted by vectors, like malaria), which is given by the prevalence. A contact resulting in an infection is called a susceptible-infected effective contact.
Infectious and non-communicable disease models have substantial similarities: both can be compartmental or agent-based (microsimulation), as well as deterministic (static transition probabilities) or stochastic (transition probabilities are random draws of a specified distribution). In any case, the decision about which model to use is determined by the scope, purpose of the analysis, and many times, the target audience for results dissemination.
In the following section I will describe compartmental, deterministic, closed-cohort models. In a closed cohort model, we assume no deaths or births, but the population remains constant over time.
Model Types
The Susceptible-Infectious (SI) Model. This is the most basic infectious disease model. It is characterized by two state variables or compartments: Susceptible (S) and Infectious (I). Here we model one transition, and once all susceptibles are infected, the epidemic is over (no deaths in this model). The transition is driven by the transmission coefficient. This is a very important concept because regardless of model type, this parameter determines the rate at which people get infected. It is usually denoted by lambda, λ, and it is the product of the infectivity or probability of transmission per contact (ρ), the contact rate at a given period (c), and the prevalence of infected (I/N; where N is the total population): λ = c * ρ * I/N. At any point in time, and for all model options, the number susceptible decreases by λ.
The Susceptible-Infectious-Recover (SIR) Model. In addition to susceptible and infected, the SIR model includes the recovered (R) compartment. R includes people that were infected and overcome the disease. The rate of transition is given by the inverse of disease duration, also known as the recovery rate (γ). Some diseases confer immunity (e.g. measles) after infection, but others do not. To capture this, a SIRS (susceptible-infected-recovered-susceptible) model would be more appropriate and allows those who don’t develop immunity to transition back to susceptible.
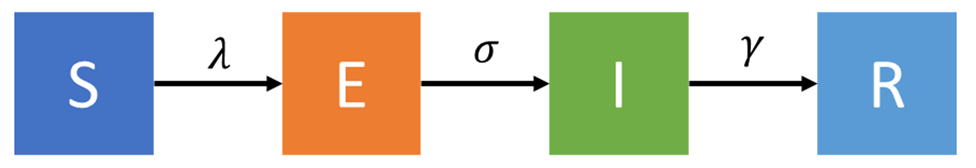
The Susceptible-Exposed-Infectious-Recovered, (SEIR) Model. This model adds an exposed (E) compartment. Exposed are all persons who have been infected but are not yet symptomatic, and more importantly, not yet infectious. Infectious persons are the only ones capable of spreading the disease, hence, an accurate count of them is very important. When using a SEIR model, the transition between S and E is given by lambda (λ) and the transition between E and I is given by the inverse of the latency or incubation period (σ).
COVID-19 Outbreak Example
I am going to simulate a COVID-19 outbreak using a SEIR model, depicted in the figure below. All parameters have been obtained from the MIDAS Network repository – an excellent and publicly available compilation of COVID-19 parameters.
Let’s model the transitions between compartments considering 1-timepoint increment:
By taking the partial derivative of these equations with respect to t, we obtain the changes in every compartment at any point in time:
With this in mind, let’s go to the R-code to see how to implement the simulation.
COVID-19 Example Results
We model an outbreak for 1 year, using the following parameters: c * ρ = 1.5, σ = 1/4.2, and γ = 1/20, for a population of 1 million where 1 persons were already infected. The following image describes the outbreak.
We can see a very steep increase in the number of infected, which peaks at 625,095 infections on the 37th day of the outbreak. As it is often pointed out, this rapid increase in cases can overload health systems, reducing the possibility of many people to access care.
How can we flatten the curve? One intervention to contain the COVID-19 pandemic was to increase the physical distance between people. The objective was to reduce the probability of an effective susceptible-infected contact. In modelling terms, this would directly reduce c * ρ and therefore λ.
The following image shows the results of reducing c * ρ to 0.6 instead of 1.5.
The peak of infection occurs later, on day 65, at a lower count as well: 550,446. This is an example of how effective behavioral changes can be to reduce the severity of an outbreak.
In this example we changed only one parameter. But one thing that amazes me about infectious disease modelling, is that (almost) every parameter driving the outbreak is susceptible to change given the right intervention. You can now use the R-code to see how variations in other parameters affect the outbreak and think about what kinds of interventions might produce such changes.
Suggested Readings
Vynnycky, E. & White, R. G. An introduction to infectious disease modelling. (Oxford University Press, 2010). BookSite
Garnett, G. An introduction to mathematical models in sexually transmitted disease epidemiology. Sex Transm Infect 78, 7–12 (2002).
Kretzschmar, M. Disease modeling for public health: added value, challenges, and institutional constraints. J Public Health Pol 41, 39–51 (2020).
On April 16, 2020, the ISPOR Student Chapter at the University of Washington hosted a webinar on how to formulate good research questions featuring Dr. Mark Bounthavong, PhD, PharmD, MPH. He discussed aspects of compelling research questions, shared his formulating process, presented best practices, and provided recommendations for students at all stages of their career.
Dr. Bounthavong is a graduate of the UW CHOICE Institute and a prolific researcher with several years of experience in HEOR. He currently serves as a health economist at the VA Health Economics Resource Center and a Research Affiliate at Stanford University, and his research interests include pharmacoeconomics, outcomes research, health economics, process and program evaluations, econometric methods, and evidence synthesis using Bayesian methods.
Our UW Student Chapter thanks Dr. Bounthavong for his insightful presentation and hopes our fellow researchers find this recording of his presentation to be a helpful resource.
Note: Dr. Bounthavong has authorized the publication of his talk in this post.